2L:10,003,782..10,003,896 [+]
Comment: 0-1 hr AEL at 24C
Comment: 1-2 hr AEL at 24C
Comment: 2-3 hr AEL at 24C
Comment: 3-4 hr AEL at 24C
Comment: 4-5 hr AEL at 24C
Comment: 5-6 hr AEL at 24C
Comment: 6-7 hr AEL at 24C
Comment: 7-8 hr AEL at 24C
Comment: 8-9 hr AEL at 24C
Comment: 9-10 hr AEL at 24C
Comment: 10-11 hr AEL at 24C
Comment: 11-12 hr AEL at 24C
Comment: 12-13 hr AEL at 24C
Comment: 14-15 hr AEL at 24C
Comment: 17-18 hr AEL at 24C
Comment: 19-20 hr AEL at 24C
Comment: 20-21 hr AEL at 24C
Comment: 22-23 hr AEL at 24C
Comment: 42-44 hr AEL 24C
Comment: 66-68 hr AEL 24C
Comment: 96 hr APF at 24C
Comment: 5 days post-eclosion
Comment: 5 days post-eclosion
Embryos were collected in one-hour windows on grape-agar plates after two two-hour prelays and aged appropriately. Embryos were dechorionated and snap frozen in liquid nitrogen.
Total RNA was isolated using the RNAdvance Tissue kit (Agencourt) and treated with DNase I. RNA quality was checked on a Bioanalyzer (Agilent). 5'-monophosphate RNA species, mainly ribosomal RNA, were depleted by TEX digest (Epicentre). Reverse transcriptase was done under conditions that promote "template switching" to preferentially add linkers to the 5' ends of 5'-complete cDNAs for second strand synthesis. 5'-capped RNA molecules were then biotinylated and pulled down to serve as templates for second strand cDNA synthesis. cDNA was PCR amplified and DNA quality was confirmed on a DNA HS Bioanalyzer chip.
Three multiplexed libraries were prepared: one for embryos (24 samples), one for larvae and pupae (10 samples) and one for adults (two samples).
Read length (bases): 101
Samples were processed as multiplexed libraries: one that pooled all 24 embryonic samples, one that pooled all 10 larval and pupal samples, and one that pooled both adult fly samples.
Read length (bases): 76
Total reads: 296,251,429
Uniquely mapped reads: 228,277,829
For peak calling, PCR duplicates, defined as reads sharing the same alignment, were removed from individual data sets. Data for the 36 different stages were combined for peak calling. To account for non-specific signal from abundant transcripts, 5' end frequency was normalized to transcript abundance as measured by the 3' portion of the paired end reads. Peaks were called by a sliding window algorithm that assesses the significance of local signal enrichment given a null distribution. Signal-enriched windows in close proximity to each other are merged into peaks, and those were subsequently trimmed at the edges down to the first base with signal.
Sequences corresponding to library identification barcodes and primers were trimmed. Reads were mapped with STAR. All uniquely mapping reads were kept. For multiply mapping reads, if all alignments started within an annotated transposon and overlapped the same gene annotation, the alignment starting in the closest transposon was selected. Transcript annotations were obtained from FlyBase (release 5.32).
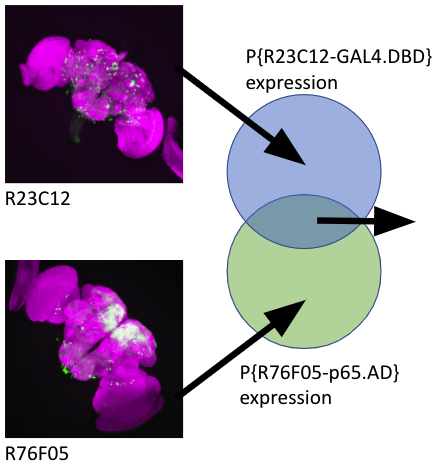

RPM=24543 (reads in TSS_region per million reads in all TSS_regions).